

Hierarchical Bayesian Hidden Markov Models for DNA Methylation
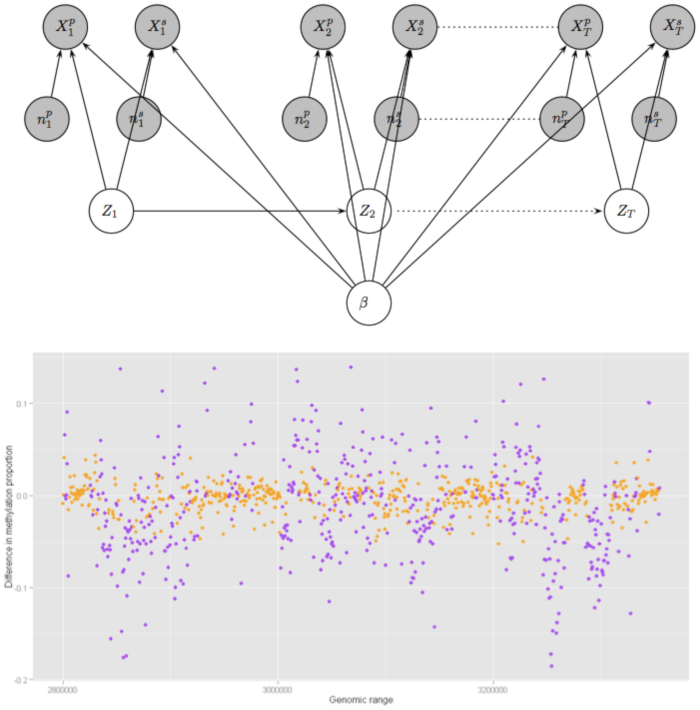
DNA Methylation is an important epigenetic mechanism for controlling gene expression, silencing or genomic imprinting in living cells. High-throughput methods, such as the sequencing of sodium bisulfite-treated DNA (BS-Seq) have been developed to study the prevalence of DNA Methylation over the whole human genome. Modelling and analysing BS-Seq data to detect differentially methylated regions (DMRs) that exhibit varying degrees of methylation in different cells or under different conditions is an important scientific problem, which poses many challenges, including adjusting for biases due to experimental artefacts, and complex dependence features within the data. We have proposed a novel methodology for predicting DMRs within a hierarchical Bayesian hidden Markov model framework, incorporating several levels of dependence between observations. Our method efficiently deals with nuisance parameters without leading to overwhelming analytical complexity and allows a principled way of building prior distributions based on partially known information, improving estimation of novel features. Application of these methods on data from a study on human aging revealed several known and novel differentially methylated regions of high biological significance in five human chromosomes.
