

Bioinformatic Workflows
Bioinformatics Workflows
Dr Ijaz’s lab specializes in developing bespoke pipelines and statistical tools for analysing genomic data in an environmental/medical context with particular focus on integrating omics datasets (amplicons and whole-genome shotgun [WGS] metagenomics, metatranscriptomics, metabolomics, metaproteomics, and population genomics) for microbial community analysis.
Some of the software that will be utilised in research grants running within the BINGO group include:
AMPLIpyth (python-based pipeline to generate operational taxonomic units [OTUs] and phylogenetic trees for 16S rRNA datasets (Meltzer 2015);
CONCOCT (software for recovering metagenome-assembled genomes [MAGs] from mixed community sample (Alneberg et al. 2014)
RVLAB (online statistical processing environment for multivariate analysis of microbial communities (Varsos et al. 2016);
NMGS (software for fitting unified theory of neutral models to microbial communities (Harris et al. 2017);
SEQENV & SEQENV-EXT (software for text mining environmental ontology terms, habitat identification from online text sources associated with genomic sequences (Ijaz et al. 2017; Sinclair et al. 2016); microbiomeSEQ (R package for microbial community analysis in an environmental context; https://github.com/umerijaz/microbiomeSeq);
NanoAmpli-Seq a workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform (Calus, Ijaz, and Pinto 2018)
PyTag An automated identification and analysis of ontological terms in gastrointestinal diseases and nutrition-related literature provides useful insights (Koci et al. 2018);
GLOBALVIEW (software for inferring associative and causal networks from time series data; transcriptomics workflow [ http://www.tinyurl.com/JCBioinformatics2], and metaproteomics workflow [ http://userweb.eng.gla.ac.uk/umer.ijaz/bioinformatics/Metaproteomics.html].
Additionally, Professor Milling and Dr Ijaz have been involved in an “IMIGPA” study to investigate treatment responses to anti-TNF antibodies in patients with psoriatic arthritis, in collaboration with Professor Anne Barton (Manchester University). In this “IMIGPA” study, which has recently completed recruitment, they have developed integrated methodologies to combine 16S sequencing data with immuno-phenotyping (Flow cytometric panels target, for example, T cells, regulatory T cells. dendritic cells, monocytes, and B cells, using antibodies specific for: CD1c, CD3, CD4, CD8, CD11c, CD14, CD15, CD16, CD19, CD25, CD27, CD45RA, CD56, CD62L. CD80, CD123, CD141, FoxP3 (i/c), and HLA-DR). These tools shown in Figure 1.
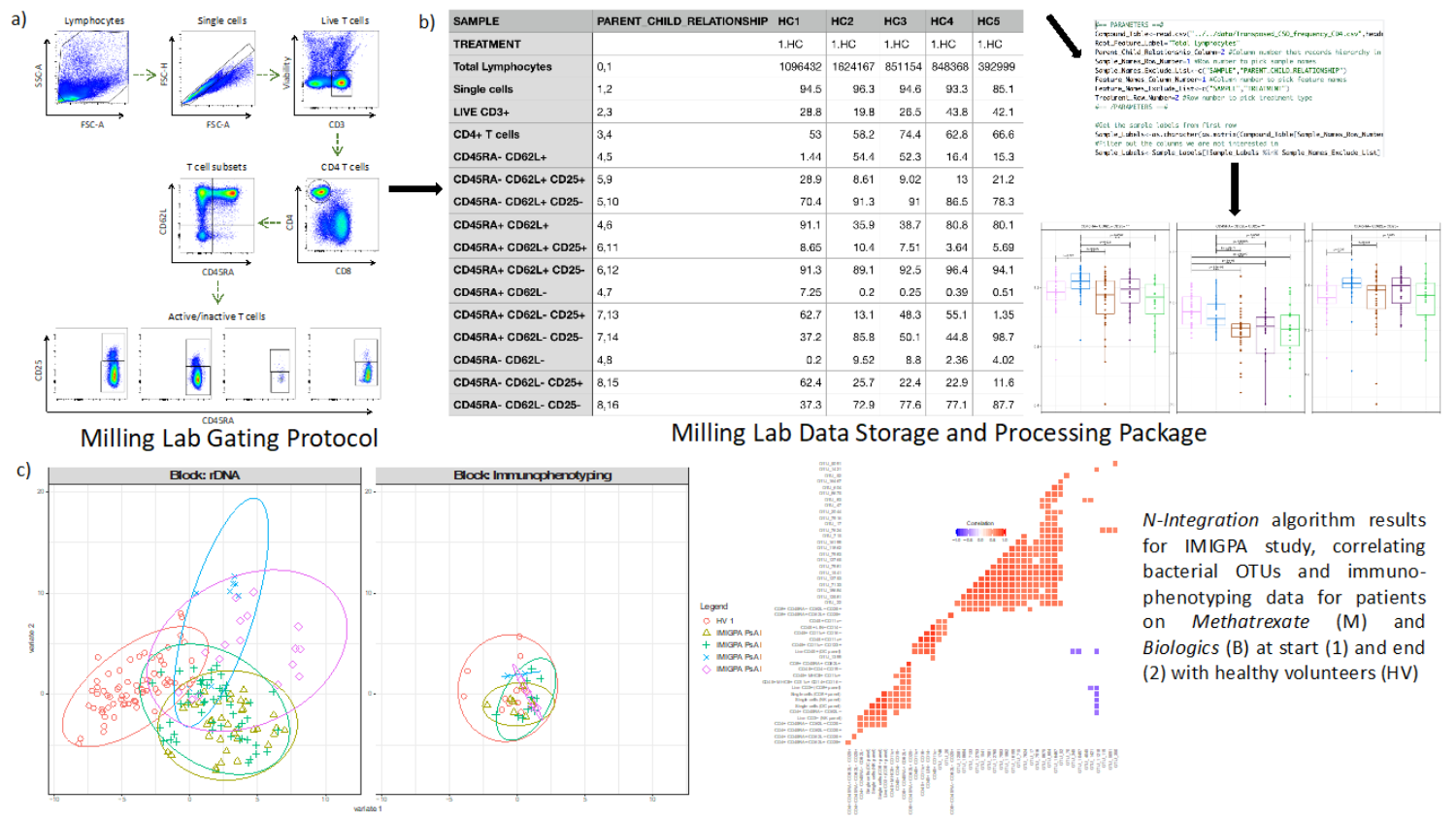 Figure 1. a) and b) show the protocol and R package developed during the IMIGPA study, c) shows bacterial OTUs correlating with immunophenotyping data (red: positive; blue: negative).
Figure 1. a) and b) show the protocol and R package developed during the IMIGPA study, c) shows bacterial OTUs correlating with immunophenotyping data (red: positive; blue: negative).
Further tools for short read amplicons include generating Amplicons Sequencing Variants (ASVs) using DADA 2 and deblur in qiime2 workflow (Bolyen et al. 2019). These are particularly useful when no appropriate threshold is available for clustering functional genes.
In addition, we will utilize third-party tools for WGS, such as:
dRep for dereplicating the genome set by identifying groups of similar genomes (Olm et al. 2017);
Recentrifuge for removing contaminants (Martí 2019);
PLASS for recovering protein residues (Steinegger, Mirdita, and Söding 2019);
Kofamscan to identify metabolic potential (KEGG orthologs) of recovered MAGs (Aramaki et al. 2019);
DESMON for extraction of strains from the MAGs (Quince et al. 2017);
abricate and
RGI for screening of antimicrobial and antibiotic genes (Jia et al. 2016);
BAGEL4 for finding bacteriocins signature (Van Heel et al. 2018);
GTDB-Tk for assigning objective taxonomic classifications to bacterial and archaeal genomes (Parks et al. 2018);
tRNAscan-SE for detecting transfer RNAs genes (Chan and Lowe 2019);
RFPlasmid [ https://github.com/aldertzomer/rfplasmid] for predicting plasmid contigs from MAGs using single copy marker genes, plasmid genes, and kmers;
NCyc [ https://github.com/qichao1984/NCyc] for fast and accurate metagenomic profiling of nitrogen cycling genes;
METABOLIC for enabling the prediction of metabolic and biogeochemical functional traits for recovered microbial genomes (Zhou et al. 2019);
Anvi’o (Eren et al. 2015) and
ATLAS (Kieser et al. 2019) pipelines for an advanced analysis and visualization platform for ‘omics data.
Some of the above methods were used in recent papers by Dr Ijaz (Sevillano et al. 2019) (Cotto et al. 2020)
Analysing bacterial isolates: reads will be mapped against fully annotated reference genomes and sample-wise reads of individual isolates will be mapped. Following this
LoFreq will be utilised to identify SNPs against the reference strain (Wilm et al. 2012). On these, we will use
popoolation2 (Kofler et al. 2011) to perform a Cochran-Mantel-Haenszel test (cmh-test.pl) for repeated tests of independence to identify significant SNPs between multiple treatment groups. Following this, and using custom scripts https://github.com/umerijaz/SNPCalling, to extract the genes sequences from reference annotations hitting the SNPs. This will enable us to use
BLAST2GO to recover GO Ontologies associated with the genes affected by the SNPs (Conesa et al. 2005). In the absence of any reference, the approach considered here will be similar to the one used in recent paper by Dr Ijaz (Dingle et al. 2019). Briefly, genomes from each isolate will be assembled using
VelvetOptimiser (Zerbino and Birney 2008). Following this, we will use
BIGSdb (Jolley and Cj Maiden 2010) to determine whether genes or nonsynonymous point mutations are present and to extract the sequences of interest, and further perform multilocus sequence type (MLST) analysis. Informed by BIGSdb, phylogenetic tree of the isolates will then be built using maximum likelihood approach such as
PhyML (Phane Guindon et al. 2005), and isolates will be annotated with “Evolutionary Distinctiveness” scores (Martyn et al. 2012). We will also annotate all the isolates using
PROKKA (Seemann 2014) to identify coding sequence (CDS) regions, and will perform bayesian concordance analysis on common CDSs between the isolate using
BUCKy (Larget et al. 2010) to estimate branch lengths in coalescent units from the quartet concordance factors. Similar to WGS step above, we can use
abricate, RGI, KofamScan, RFPlasmid and BAGEL4 to identify additional features.
In terms of third-party statistical tools (those not covered in Dr Ijaz’s software), we will use: (a) phylogeny-aware methods to identify stochastic (phylogenetic overdispersion) or deterministic (phylogenetic clustering) factors affecting microbiome diversity and structure all as implemented in the R package
PhyloMeasures (Tsirogiannis and Sandel 2016) (b) Clarke and Ainsworth’s BIOENV and BVSTEP (Clarke and Ainsworth 1993) routines for comparison of dissimilarity measure on the multivariate statistical tables (generated from above software) to identify the main distinguishing features. (c) Random Forest Classifiers trained on multiples conditions on the tables (from the above pipelines) to assign importance measures for each feature, such as mean decrease gini and mean decrease accuracy (e. g Species, KEGGs, RNA transcripts, Proteins). Integration of taxonomic information will enable measures to be propagated up taxonomic levels to identify important clades and/or functions. (d) N-and P- integration algorithms such as MINT, and DIABLO available in mixOmics [http://mixomics.org/] to classify and discriminate features that correlate across multiple datasets (genomic dataset, metabolomics dataset, SCFAs, flow cytometry data and any other clinical metadata; see Dr Ijaz’s recent work for integrating metabolomics profiles and microbial community data (Gauchotte-Lindsay et al. 2019)) (e) Pairwise enrichment analyses comparing different treatment groups using either negative binomial GLM fitting (DESeq2 (Love, Huber, and Anders 2014)); non-parametric tests such as Kruskal-Wallis, with appropriate normalisation; Kernel based approaches for metabolomics data (LC-MS, GC-MS) [(Zhan, Patterson, and Ghosh 2015)]; ANCOM differential analysis [(Mandal et al. 2015)]; balances or log ratio as an alternative technique to investigate microbial composition [https://docs.qiime2.org/2019.7/tutorials/gneiss/] (f) Longitudinal statistics for microbial communities include: temporal stability and volatility analysis for indicating periods of disruption and abnormal events (Bokulich et al. 2018) non-parametric microbial interdependence test (NMIT) to determine longitudinal sample similarity [(Zhang et al. 2017)]; Negative Binomial Smoothing Spline ANOVA to identify time intervals when OTUs/MAGs/Pathways become significant [(Metwally et al. 2018)]; and microbial maturity index from a regression model trained on feature data to predict a subject’s age as a function of microbiota composition (Subramanian et al. 2014)
References
Alneberg, Johannes et al. 2014. “Binning Metagenomic Contigs by Coverage and Composition.” Nature Methods 11(11): 1144–46.
Aramaki, Takuya et al. 2019. “KofamKOALA: KEGG Ortholog Assignment Based on Profile HMM and Adaptive Score Threshold.” Bioinformatics btz859: 602110.
Bokulich, Nicholas A. et al. 2018. “Q2-Longitudinal: Longitudinal and Paired-Sample Analyses of Microbiome Data.” mSystems 3(6).
Bolyen, Evan et al. 2019. “Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2.” Nature Biotechnology 37(8): 852–57.
Calus, Szymon T, Umer Zeeshan Ijaz, and Ameet J Pinto. 2018. “NanoAmpli-Seq: A Workflow for Amplicon Sequencing for Mixed Microbial Communities on the Nanopore Sequencing Platform” GigaScience 7(12). https://academic.oup.com/gigascience/article/7/12/giy140/5202451 (March 11, 2020).
Chan, Patricia P., and Todd M. Lowe. 2019. “TRNAscan-SE: Searching for TRNA Genes in Genomic Sequences.” In Methods in Molecular Biology, Humana Press Inc., 1–14.
Clarke, KR, and M Ainsworth. 1993. “A Method of Linking Multivariate Community Structure to Environmental Variables.” Marine Ecology Progress Series 92: 205–19. https://www.int-res.com/articles/meps/92/m092p205.pdf (September 30, 2019).
Conesa, Ana et al. 2005. “Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research.” Bioinformatics 21(18): 3674–76. http://www.blast2go.de (March 11, 2020).
Cotto, Irmarie et al. 2020. “Long Solids Retention Times and Attached Growth Phase Favor Prevalence of Comammox Bacteria in Nitrogen Removal Systems.” Water Research 169: 115268.
Dingle, Kate E. et al. 2019. “A Role for Tetracycline Selection in Recent Evolution of Agriculture-Associated Clostridium Difficile Pcr Ribotype 078.” mBio 10(2).
Eren, A. Murat et al. 2015. “Anvi’o: An Advanced Analysis and Visualization Platformfor ’omics Data.” PeerJ 2015(10): e1319.
Gauchotte-Lindsay, Caroline, Thomas J. Aspray, Mara Knapp, and Umer Z. Ijaz. 2019. “Systems Biology Approach to Elucidation of Contaminant Biodegradation in Complex Samples – Integration of High-Resolution Analytical and Molecular Tools.” Faraday Discussions 218(0): 481–504. http://xlink.rsc.org/?DOI=C9FD00020H (September 30, 2019).
Harris, Keith et al. 2017. “Linking Statistical and Ecological Theory: Hubbell’s Unified Neutral Theory of Biodiversity as a Hierarchical Dirichlet Process:” In Proceedings of the IEEE, Institute of Electrical and Electronics Engineers Inc., 516–29.
Van Heel, Auke J et al. 2018. “BAGEL4: A User-Friendly Web Server to Thoroughly Mine RiPPs and Bacteriocins.” Nucleic Acids Research 46. http://bagel4.molgenrug.nl/blast.php (March 11, 2020).
Ijaz, Ali Z. et al. 2017. “Extending SEQenv: A Taxa-Centric Approach to Environmental Annotations of 16S RDNA Sequences.” PeerJ 2017(10): e3827.
Jia, Baofeng et al. 2016. “CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database.” Nucleic Acids Research 45(D1): D566–D573. https://academic.oup.com/nar/article/45/D1/D566/2333912 (March 11, 2020).
Jolley, Keith A, and Martin Cj Maiden. 2010. “BIGSdb: Scalable Analysis of Bacterial Genome Variation at the Population Level.” BMC bioinformatics 11. http://pubmlst.org/software/database/bigsdb/. (March 11, 2020).
Kieser, Silas et al. 2019. “ATLAS: A Snakemake Workflow for Assembly, Annotation, and Genomic Binning of Metagenome Sequence Data.” bioRxiv: 737528. https://www.biorxiv.org/content/10.1101/737528v1 (March 11, 2020).
Koci, Orges et al. 2018. “An Automated Identification and Analysis of Ontological Terms in Gastrointestinal Diseases and Nutrition-Related Literature Provides Useful Insights.” PeerJ 6(7): e5047. https://peerj.com/articles/5047 (September 21, 2018).
Kofler, Robert, Ram Vinay Pandey, Christian Schlötterer, and Jeffrey Barrett. 2011. “PoPoolation2: Identifying Differentiation between Populations Using Sequencing of Pooled DNA Samples (Pool-Seq).” Bioinformatics 27(24): 3435–36 (March 11, 2020).
Larget, Bret R, Satish K Kotha, Colin N Dewey, and Cécile Ané. 2010. “BUCKy: Gene Tree/Species Tree Reconciliation with Bayesian Concordance Analysis.” Bioinfomatics 26(22): 2910–11. https://academic.oup.com/bioinformatics/article-abstract/26/22/2910/227750 (March 11, 2020).
Love, Michael I, Wolfgang Huber, and Simon Anders. 2014. “Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2.” Genome Biology 15(12): 550. http://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 (September 30, 2019).
Mandal, Siddhartha et al. 2015. “Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition.” Microbial Ecology in Health & Disease 26(0).
Martí, Jose Manuel. 2019. “Recentrifuge: Robust Comparative Analysis and Contamination Removal for Metagenomics.” PLoS Computational Biology 15(4).
Martyn, Iain et al. 2012. “Computing Evolutionary Distinctiveness Indices in Large Scale Analysis.” Algorithms for Molecular Biology 7(1): 6. http://almob.biomedcentral.com/articles/10.1186/1748-7188-7-6 (March 11, 2020).
Meltzer, David Jacob. 2015. AMPLIpyth: A Python Pipeline for Amplicon Processing.
Metwally, Ahmed A. et al. 2018. “MetaLonDA: A Flexible R Package for Identifying Time Intervals of Differentially Abundant Features in Metagenomic Longitudinal Studies.” Microbiome 6(1): 32. https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-018-0402-y (March 11, 2020).
Olm, Matthew R., Christopher T. Brown, Brandon Brooks, and Jillian F. Banfield. 2017. “DRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication.” ISME Journal 11(12): 2864–68.
Parks, Donovan H. et al. 2018. “A Standardized Bacterial Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life.” Nature Biotechnology 36(10): 996.
Phane Guindon, Sté, Franck Lethiec, Patrice Duroux, and Olivier Gascuel. 2005. “PHYML Online-a Web Server for Fast Maximum Likelihood-Based Phylogenetic Inference.” Nucleic Acids Research 33: W557–W559. https://academic.oup.com/nar/article-abstract/33/suppl_2/W557/2505422 (March 11, 2020).
Quince, Christopher et al. 2017. “DESMAN: A New Tool for de Novo Extraction of Strains from Metagenomes.” Genome Biology 18(1): 181. http://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1309-9 (March 11, 2020).
Seemann, Torsten. 2014. “Prokka: Rapid Prokaryotic Genome Annotation.” Bioinformatics 30(14): 2068–69. https://academic.oup.com/bioinformatics/article-abstract/30/14/2068/2390517 (March 11, 2020).
Sevillano, Maria et al. 2019. “Disinfectant Residuals in Drinking Water Systems Select for Mycobacterial Populations with Intrinsic Antimicrobial Resistance.” bioRxiv: 675561.
Sinclair, Lucas et al. 2016. “Seqenv: Linking Sequences to Environments through Text Mining.” PeerJ 2016(12): e2690.
Steinegger, Martin, Milot Mirdita, and Johannes Söding. 2019. “Protein-Level Assembly Increases Protein Sequence Recovery from Metagenomic Samples Manyfold.” Nature Methods 16(7): 603–6.
Subramanian, Sathish et al. 2014. “Persistent Gut Microbiota Immaturity in Malnourished Bangladeshi Children.” Nature 510(7505): 417–21.
Tsirogiannis, Constantinos, and Brody Sandel. 2016. “PhyloMeasures: A Package for Computing Phylogenetic Biodiversity Measures and Their Statistical Moments.” Ecography 39(7): 709–14. http://doi.wiley.com/10.1111/ecog.01814 (March 11, 2020).
Varsos, Constantinos et al. 2016. “Optimized R Functions for Analysis of Ecological Community Data Using the R Virtual Laboratory (RvLab).” Biodiversity Data Journal 4: e8357. http://bdj.pensoft.net/articles.php?id=8357 (September 21, 2018).
Wilm, Andreas et al. 2012. “LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets.” Nucleic Acids Research 40(22): 11189–11201. http://sourceforge.net/projects/lofreq/. (March 11, 2020).
Zerbino, Daniel R, and Ewan Birney. 2008. “Velvet: Algorithms for de Novo Short Read Assembly Using de Bruijn Graphs.” Genome research 18(5): 821–29. www.genome.org/cgi/doi/10.1101/gr.074492.107 (March 11, 2020).
Zhan, Xiang, Andrew D. Patterson, and Debashis Ghosh. 2015. “Kernel Approaches for Differential Expression Analysis of Mass Spectrometry-Based Metabolomics Data.” BMC Bioinformatics 16(1): 77. http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-015-0506-3 (March 11, 2020).
Zhang, Yilong, Sung Won Han, Laura M. Cox, and Huilin Li. 2017. “A Multivariate Distance-Based Analytic Framework for Microbial Interdependence Association Test in Longitudinal Study.” Genetic Epidemiology 41(8): 769–78.
Zhou, Zhichao et al. 2019. “METABOLIC: A Scalable High-Throughput Metabolic and Biogeochemical Functional Trait Profiler Based on Microbial Genomes.” bioRxiv: 761643. https://www.biorxiv.org/content/10.1101/761643v1 (March 11, 2020).